
DNA Repair Laboratory
Introduction
Investigations into the development of DNA damage and the molecular mechanisms for its repair in the genome of mammalian cells are at the forefront of our work. In particular, we use rodent models and clinical samples to investigate the significance of these processes for the individual cancer risk and for the development of therapy resistance in tumor diseases.
We are also interested in the complex response of cells after DNA damage by cytostatic drugs and the interaction with other functions such as cell cycle regulation and apoptosis.

Jürgen Thomale
Im Ruhestand
(Retired)
Research projects from the DNA Repair Laboratory
Regulation of DNA repair functions in mammalian cells
The ability of mammalian cells to deal with DNA lesions varies according to cell type and differentiation. In humans, significant inter-individual differences have also been observed. When analyzing normal and malignant hematopoietic cells, we look for common regulatory patterns within the “DNA damage response network” and try to identify rate-limiting steps along the major repair pathways. Functional data from leukemic cells suggest that the tight control and fine-tuning of this complex network is (partially) abolished in physiological cells during carcinogenesis. Loosened regulation of damage response genes could in turn facilitate the outgrowth of resistant cell clones in vivo under the selection pressure of DNA-reactive anticancer drugs.
Using laboratory-based immunoanalytical techniques, we can visualize and measure drug-induced DNA damage at single-cell resolution and thus analyze such processes in model systems and in clinical samples. Our research focuses primarily on the role of DNA repair mechanisms for individual cancer risk and for the chemosensitivity of e.g. hematopoietic stem and progenitor cells.
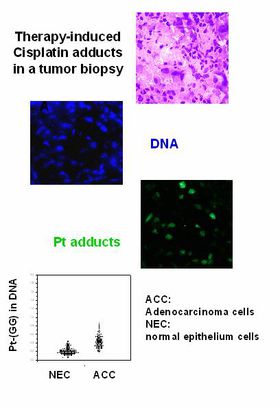
Global genomic vs. gene-specific repair: What is important for the efficacy of cancer drugs that interact with DNA?
Depending on their actual location in the genome of mammalian cells, structural lesions can persist for different lengths of time. This “toposelectivity” of DNA repair is due to specialized mechanisms that only take care of active genes or their transcribed strand (transcription-coupled repair; TCR). These specific features have a major influence on the mutagenic and cytotoxic potency of DNA-damaging substances. However, the relative importance of “toposelective” versus global genomic repair for the chemosensitivity of tumor cells is largely unknown.
Experiments comparing wild-type and TCR-deficient mice have shown an altered toxicity profile for a number of anticancer drugs in “knockout” animals. Thus, key components of these signaling pathways can be identified as potential targets for pharmacological modulation of drug resistance. Our group has developed monoclonal antibodies for drug-induced DNA lesions to monitor their sequence-specific distribution and elimination in normal and tumor tissues.
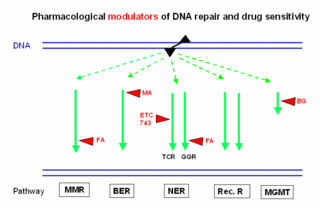
Interference of signal-modulating drugs with cellular processing of DNA damage
Cancer treatment regimens often contain molecules that modulate critical regulatory functions in tumor cells. Some of these “signaling modulators” show a pronounced enhancement of effect when combined with DNA-responsive cytostatic drugs. The interface for this synergism probably lies in the complex network that coordinates the cellular response to DNA injury. In collaboration with the group of Dr. Moritz (Experimental Hematology, Tumor Clinic), we are investigating the interplay of signal modulation and DNA repair functions in cell lines and in primary tumor cells in order to gain information for rational drug combinations.
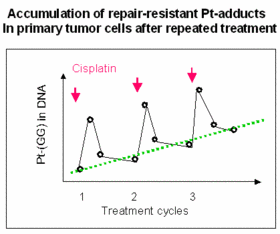
Cellular and molecular parameters that determine the tissue-specific toxicity of drugs
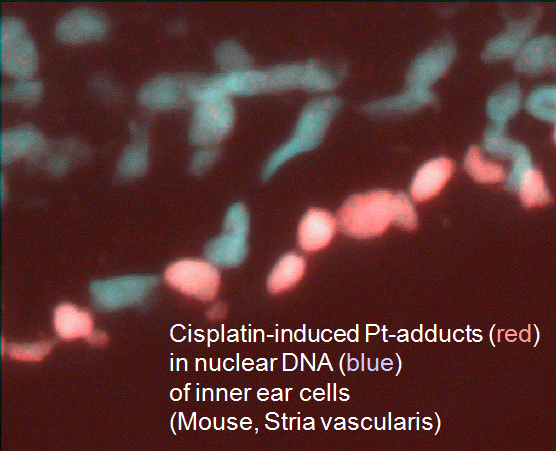
Anticancer drugs often cause a typical spectrum of toxic side effects, limiting the doses that can be used. This toxicity may be related to increased formation or prolonged persistence of drug-induced DNA lesions in critical cells of the affected tissue. As a paradigm, we analyze the molecular and cellular mechanisms underlying severe cytotoxicity e.g. in the inner ear and peripheral nervous system after cisplatin chemotherapy (collaboration with colleagues in neurology and ENT). Using normal and repair transgenic mouse models, we have shown that the risk of cisplatin-induced polyneuropathy is related to the activity of a specific DNA repair pathway in the dorsal root ganglion cells.
On the other hand, the occurrence of deafness after cisplatin treatment coincides with a very high accumulation of drug-induced adducts in a small number of specific cells in the cochlea. We are trying to identify relevant pharmacodynamic parameters (such as membrane transporters) and the cascade of sequential events leading to loss of function. In addition, the established mouse models can be used to develop and validate new protection strategies.