NG Rudner
Research focus
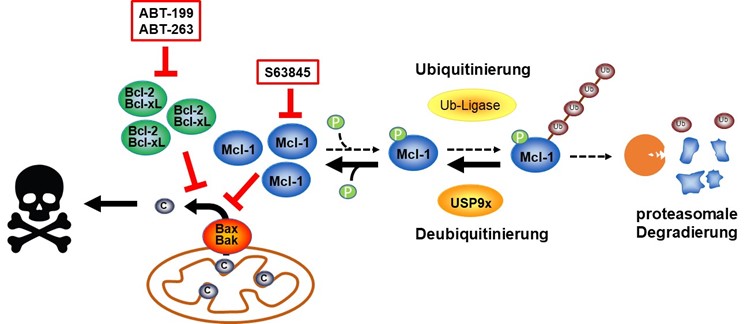
The success of anti-cancer therapies depends on how well cancer initiate cell death. As a specialised form of cell death, apoptosis (programmed cell death) contributes to cytotoxicity in response to therapeutical treatment. Ionising radiation, commonly used in anti-cancer therapies, induces apoptosis via the intrinsic apoptosis signalling pathway, which is controlled by members of the Bcl-2 protein family at the mitochondria. The anti-apoptotic members of the Bcl-2 protein family block apoptosis and thus prevent the tumour from responding to therapy, while pro-apoptotic members of the Bcl-2 protein family have the opposite effect. During cancerogenesis, tumour cells often upregulate anti-apoptotic and downregulate pro-apoptotic proteins. This is one reason why tumour cells respond very poorly to therapies, particularly at late stages of tumour progression. One focus of this research group is the analysis of regulatory mechanisms of Bcl-2 proteins during tumour progression and therapy response with the aim of intervening in these mechanisms to improve the response of tumours to therapy.
Ionising radiation causes damage to cellular molecules by ionisation. Yet, radiotherapy works better in tumour tissue that is well supplied with oxygen, as ionising radiation also interacts with oxygen to form reactive oxygen species (ROS). ROS react in turn with cellular macromolecules and cause additional damage. However, tumours display hypoxic areas insufficiently supplied with oxygen. Within the cell, mitochondria are the main source of ROS production, which can be increased even further upon damage of mitochondria. Tumour cells protect themselves particularly well against oxidative damage caused by ROS by increasing their anti-oxidative defence to neutralise ROS.
Another aim of this research group is the investigation of protective mechanism against oxidative damage in normoxic and hypoxic tumour cells in order to improve radiotherapy by increasing ROS formation.

PD Dr. rer. nat.
Justine Rudner
Nachwuchsgruppenleiterin
(Junior group leader)
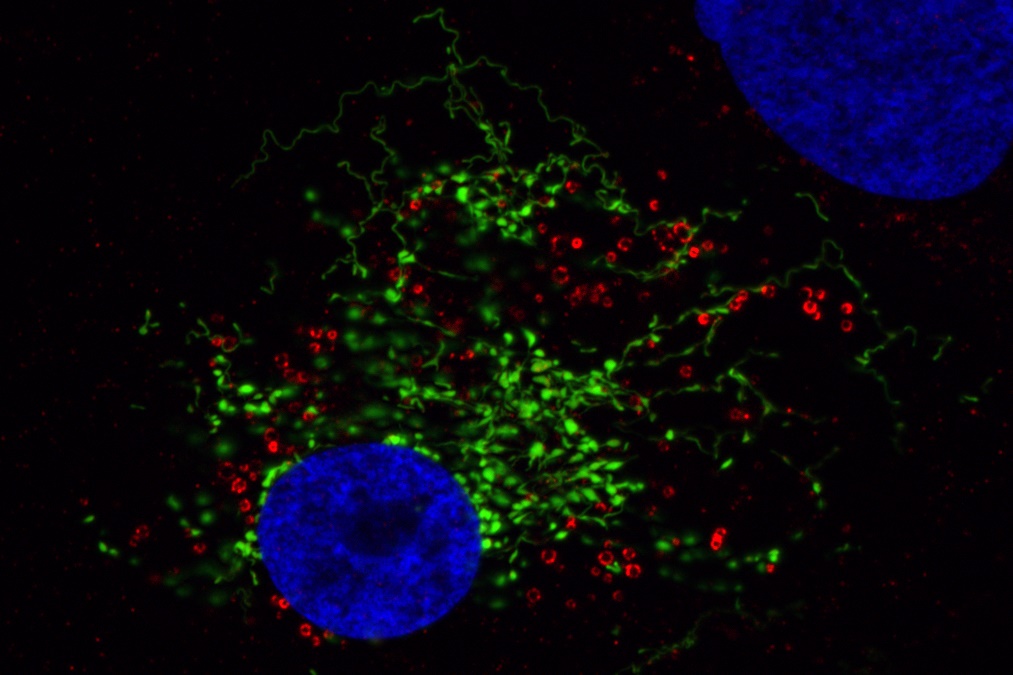
Visualisation of oxidative damage at mitochondria using MitoTimer (a green fluorescent protein targeted to mitochondria that becomes red fluorescent upon oxidation). Mitochondria fuse and divide permanently to form a network in the cell. This enables the exchange of mitochondrial content between individual mitochondria. Generation of oxidative stress by addition of dihydroartemisinine ( a chemical compound that facilitates generation of ROS) results in mitochondrial fragmentation followed by oxidative damage to the mitochondria.