NG Rudner –
Mitochondrien und
Therapieresistenz
Forschungsschwerpunkt
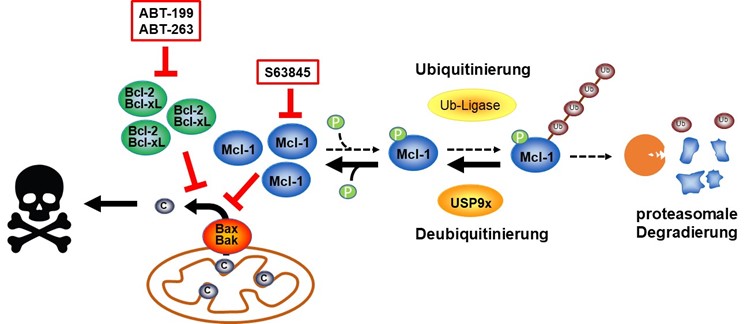
Der Erfolg der gegen den Krebs gerichteten Therapien hängt mitunter davon ab, wie gut die Krebszellen den Zelltod einleiten. Als eine spezielle Form des Zelltods trägt die Apoptose (programmierter Zelltod) zu der durch die Therapie induzierte Zytotoxizität bei. Ionisierende Strahlen, die häufig in Krebstherapien verwendet werden, induzieren Apoptose über den intrinsischen Apoptose-Signalweg, der an den Mitochondrien durch die Mitglieder der Bcl-2-Proteinfamilie kontrolliert wird. Die anti-apoptotischen Mitglieder der Bcl-2-Proteinfamilie blockieren Apoptose und verhindern so das Ansprechen des Tumors auf die Therapie, während pro-apoptotische Mitglieder der Bcl-2-Proteinfamilie den gegenteiligen Effekt haben. Tumorzellen regulieren häufig anti-apoptotische Proteine herauf, während sie pro-apoptotische Protein herunterregulieren. Dies ist ein Grund weshalb Tumorzellen sehr schlecht auf Therapien reagieren. Ein Fokus dieser Arbeitsgruppe liegt auf der Analyse der regulierenden Mechanismen der Bcl-2-Proteine im Rahmen der Tumorprogression und Therapieantwort mit dem Ziel, in diese Mechanismen einzugreifen, um das Ansprechen der Tumoren auf eine Therapie zu verbessern.
Die Strahlentherapie wirkt besser in Tumorgeweben, die gut mit Sauerstoff versorgt sind, da ionisierende Strahlung mit Sauerstoff reagiert und reaktive Sauerstoffspezies bildet. Diese reaktiven Sauerstoffspezies interagieren mit zellulären Makromolekülen und schädigen diese zusätzlich. Tumorzellen weisen jedoch häufig Areale auf, die mit Sauerstoff unterversorgt sind. Intrazellulär sind Mitochondrien die Hauptquelle der reaktiven Sauerstoffspeziesproduktion, die nach Schädigung der Mitochondrien sogar noch weiter gesteigert werden kann. Tumorzellen können sich besonders gut gegen die durch die reaktiven Sauerstoffspezies verursachten oxidative Schäden schützen, indem sie u.a. verstärkt Moleküle bilden, die die reaktiven Sauerstoffspezies neutralisieren. Ein weiteres Ziel dieser Arbeitsgruppe ist die Untersuchung der protektiven Mechanismen gegenüber oxidativen Schäden der Tumorzellen in normoxischen und hypoxischen Arealen, um diese Mechanismen gezielt zu manipulieren, um die Bildung der reaktiven Sauerstoffspezies zu steigern und so die Strahlentherapie zu verbessern.

PD Dr. rer. nat.
Justine Rudner
Nachwuchsgruppenleiterin
(Junior group leader)
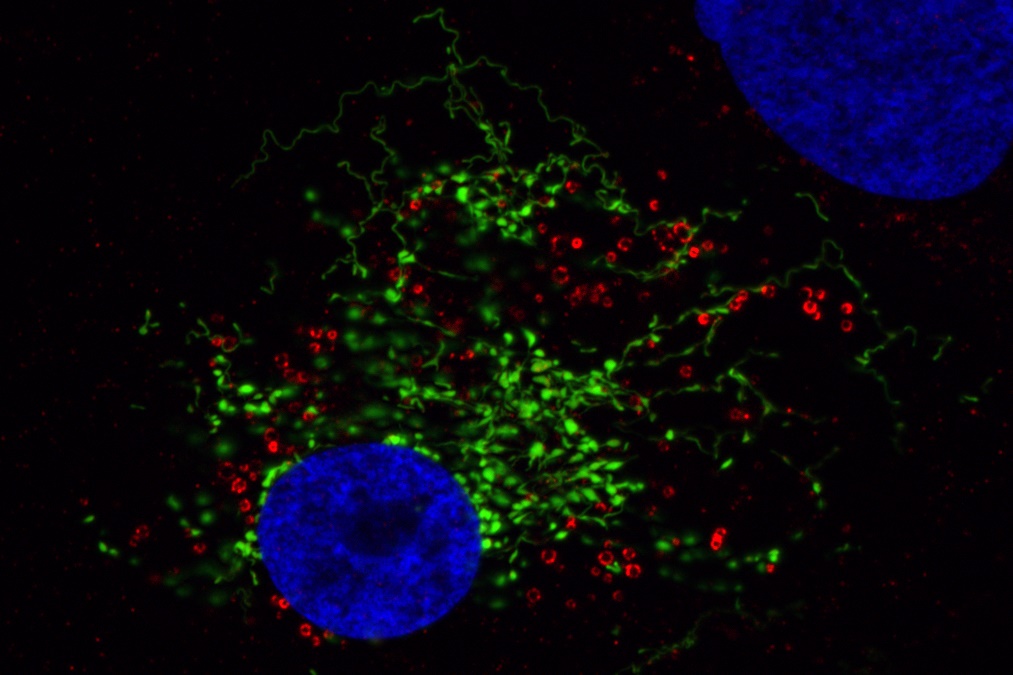
Darstellung der Schädigung der Mitochondrien mithilfe von MitoTimer (grün-fluoreszierendes Protein wird nach Oxidation zum rot-fluoreszierendem Protein). Mitochondrien fusionieren und teilen sich permanent. Dies ermöglicht den Austausch von mitochondrialem Material zwischen den einzelnen Mitochondrien. Die Erzeugung vom oxidative Stress durch Zugabe von Dihydroartemisinin (eine chemische Verbindung, die reaktiven Sauerstoffspezies produziert und bereits zur Behandlung von Malaria eingesetzt wird) resultiert nach einiger Zeit in mitochondrialer Fragmentierung, gefolgt von einer oxidativen Schädigung der Mitochondrien (Mitochondrien werden zunehmend rot).